Заболевание кошачий крик. Кошачий крик. история редкой девочки с синдромом лежёна. Внешние признаки синдрома крика кошки
Синдром кошачьего крика – редкая врожденная проблема, формирующаяся вследствие возникновения мутации в коротком плече пятой хромосомы. Частота встречаемости заболевания не превышает одного случая на 45 тыс. новорожденных. Симптомы включают в себя характерный плач, возникающий из-за деформации гортани, изменение строения черепа, отставание в развитии. Синдром Лежена, названный в честь генетика, впервые описавшего его в 1963 году, зачастую сочетается с другими врожденными патологиями. Лечение носит симптоматический характер и направлено на предупреждение развития осложнений. Важную роль играют также занятия с дефектологом и устранение речевых нарушений.
Причины возникновения синдрома кошачьего крика
Заболевание является врожденным и характеризуется способностью к наследованию. Это означает, что патология имеет генетическую природу. Синдром Лежена формируется в результате мутации, приводящей к изменению строения 5-й хромосомы, а именно ее короткого плеча. Эта структура либо укорачивается, либо теряется полностью. Встречается и мозаичная форма болезни. Среди причин возникновения синдрома кошачьего крика выделяют воздействие следующих неблагоприятных факторов:
- Значение имеет возраст будущей матери. По мере старения женского организма повышается риск развития генетических мутаций, способных приводить к формированию врожденных заболеваний. Данная закономерность прослеживается у представительниц прекрасного пола старше 45 лет.
- Употребление алкоголя и курение в подростковом возрасте и во время беременности. Вредные привычки оказывают негативное влияние как на организм матери, так и на плод.
- Заражение вирусными и бактериальными инфекциями в период вынашивания ребенка. Ряд болезнетворных агентов способны изменять процесс развития малыша, что приводит к формированию различных нарушений.
- Воздействие радиации – один из ключевых факторов в возникновении спонтанных мутаций. Излучение способно незаметно проникать в клетки человеческого организма и негативно сказываться на структуре генетического материала.
Характерные симптомы патологии
В медицине принято выделять несколько основных клинических признаков синдрома кошачьего крика:
- Характерный плач новорожденного. Этот симптом регистрируется сразу после появления ребенка на свет. Издаваемые звуки напоминают мяуканье, из-за чего заболевание и получило свое название. Появление данного признака связано с изменением структуры надгортанника, хрящей и слизистой оболочки в области глотки. Синдром мяуканья исчезает к годовалому возрасту.
- Деформации черепа. Наиболее распространенной является микроцефалия, то есть уменьшение размеров головы ребенка. Подобное нарушение провоцирует развитие неврологической симптоматики в будущем, а также является органической основой умственной отсталости.
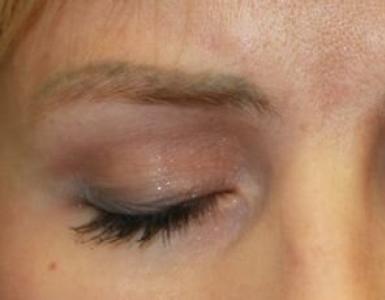
- Изменение расположения и разреза глаз. Такое проявления связано с деформацией структуры черепа. Существует несколько вариаций данного клинического признака. У некоторых детей встречается антимонголоидный вариант нарушения, другие страдают от гипертелоризма – увеличения расстояния между глазами (фото представлено ниже).
- Малыши с синдромом Лежена рождаются со сниженной массой тела и мышечной слабостью. В дальнейшем они значительно отстают от сверстников в развитии как в физическом, так и в умственном.
- Зачастую у малышей недоразвита нижняя челюсть. Это не только приводит к возникновению характерных черт лица, но и усугубляет недоразвитость ребенка, поскольку значительно затрудняет процесс грудного вскармливания.
Сопутствующие заболевания
Особенность врожденных патологий в том, что они зачастую сочетаются между собой. При синдроме Лежена, кроме характерных изменений структуры черепа и отставания в развитии, диагностируются и другие поражения. У детей с заболеванием высок риск возникновения пороков сердца, значительно ухудшающих прогноз недуга. Нарушается также работа желудочно-кишечного тракта, что сопровождается частыми запорами. Малыши страдают от дефектов костно-мышечного аппарата, например, изменения строения кистей рук и искривления позвоночника. Зачастую страдают также почки и печень, формируются тяжелые нарушения метаболизма. Распространенной проблемой при синдроме кошачьего крика являются грыжи, требующие хирургического лечения.
Диагностика
Подтверждение наличия заболевания у новорожденного проводится как на основании клинических признаков, так и при помощи специальных тестов. Деформации черепа, характерный плач и отставание в развитии дают основание предположить проблему. Основу диагностики синдрома кошачьего крика составляет определение кариотипа как малыша, так и родителей. Для подтверждения наличия отклонения в пренатальном периоде делается УЗИ.
В ходе исследования выявляются признаки хромосомных делеций: изменение количества околоплодных вод, деформация костей черепа, пороки сердца. Однако на этом этапе поставить точный диагноз не удается. После рождения ребенка, кроме генетических анализов, используется ЭХО, рентген, а также гематологические тесты, позволяющие оценить функцию внутренних органов.
Пренатальная диагностика не слишком распространена при выявлении заболевания. Например, изучение структуры хромосом плода практически не используется. При этом в ряде случаев проводится исследование ребенка при помощи ультразвука. Косвенными показателями, указывающими на формирование патологии, являются два признака: не иммунная водянка плода, а также вентрикуломегалия, то есть увеличение объемов мозговых желудочков.
К числу редких симптомов заболевания в антенатальном периоде относят формирование кист сосудистых сплетений плаценты, хотя и это проявление не является специфическим. Есть также данные об обнаружении в ходе УЗИ микроцефалии и гипоплазии мозжечка.
Несмотря на то что генетический анализ является самым достоверным методом при постановке диагноза, как в пренатальный период, так и после рождения ребенка, подобные тесты используются довольно редко. Это связано с тем, что отклонение в строении 5-й хромосомы не всегда сопровождается возникновением синдрома кошачьего крика.
Подобные исследования проводятся врачами только в тех случаях, когда у пациента отмечается наличие специфических клинических признаков, позволяющих заподозрить данную проблему. В таких случаях осуществление генетического анализа используется с целью дифференциации заболевания от других дефектов со сходной симптоматикой.

Существующие методы лечения
Специфических мер борьбы с синдромом кошачьего крика не разработано. Если у ребенка выявляются сопутствующие заболевания, например пороки клапанного аппарата сердца, проводится их оперативная коррекция. К хирургическим техникам прибегают также при наличии у малыша врожденных грыж. В ряде случаев, когда пациенты страдают от недостаточности функции почек и печени, проводится терапия в зависимости от стадии процесса. Лечение при заболевании паллиативное, то есть носит симптоматический характер, а также направлено на профилактику осложнений.
Важным этапом терапии является стимуляция нормального умственного и физического развития ребенка. Для этого применяется физиотерапия, основанная на использовании воздействия тепла, и занятия в бассейне. Хорошие результаты показывают также массажные техники и лечебная физкультура, направленная на улучшение тонуса мышц. Детям требуется психотерапия, позволяющая скорректировать отклонения в функции высшей нервной деятельности.
Даже при адекватном лечении дети с синдромом Лежена не могут обслуживать себя. Они нуждаются в постоянном надзоре и уходе, в том числе и в случаях, когда пациенты доживают до преклонного возраста.
Прогноз и профилактика
Исход заболевания определяется выраженностью изменений, а также наличием сопутствующих патологий. У пациентов повышен риск развития осложнений со стороны дыхательной и мочевыделительной систем, а также имеются значительные трудности в сфере социальных взаимодействий. В целом прогноз при синдроме Лежена стремится к благоприятному. Благодаря современным методам диагностики и лечения поражений различных органов, больным удается дожить до 40–50 лет.
Профилактика патологии сводится к правильному планированию и ведению беременности. Будущим родителям, семейный анамнез которых осложнен случаями выявления хромосомных мутаций, рекомендуется проходить генетические тесты. Женщинам в период вынашивания ребенка следует избегать воздействия стрессов, правильно питаться, а также отказаться от вредных привычек. Специфических методов профилактики проблемы не разработано.
Кариотип 46 ХХ или ХУ, 5р-. Соотношение полов - МI: ЖI. Частота - 1:40 - 50 тыс. новорожденных.
Специфический плач, напоминающий «кошачье мяуканье»;
Низкая масса при рождении;
Умственное или физическое недоразвитие;
Микроцефалия, птоз, низкое расположение и деформация ушных раковин, кожные складки впереди уха, гипертелоризм, эпикант, антимонголоидный разрез глаз, лунообразное лицо;
Мышечная гипотония;
Врожденные пороки развития, грыжи, расхождение прямых мышц живота;
Плоскостопие, «обезьянья складка»;
Такие признаки, как «кошачий крик», мышечная гипотония, лунообразное лицо, в большинстве случаев полностью изчезают с возрастом. Но большинство детей умирает в раннем возрасте.
2. Синдром Прадера-Вилли (у мужчин) и синдром Ангельмана (у женщин) (q87.1)
Кариотип 46 ХХ или ХУ, 15р-.
Клиника - мышечная гипотония, гипогонадизм, ожирение, умственная отсталость, маленькие кисти и стопы, микроцефалия, высокое арковидное небо, кариес, микродонтия, гипоплазия ушных раковин, сколиоз, синдактилия, поперечная ладонная складка, нарушение координации движений, судороги, сахарный диабет.
Дупликация - удвоение хромосомы. Тандемная дупликация - возникновение соседнего плеча добавочной хромосомы от центромеры хромосомы. Механизм аберрации:
Вследствие увеличения того или иного сегмента хромосомы;
Вследствие несбалансированных транслокаций.
3. Синдром 9 р+ (синдром Реторе)(Q92.9).
Кариотип 46 ХХ или ХУ, 9р+. По частоте встречаемости среди детей-олигофренов занимает 2 место после болезни Дауна.
Клинико-морфологические признаки:
Пренатальная умеренная гипоплазия, задержка роста;
Микроцефалия, антимонголоидный разрез глаз;
Умственная отсталость;
Гиперплазия 3 и 4 фаланг пальцев;
Пороки развития внутренних органов (сердца и почек).
Прогноз жизни благоприятный при отсутствии патологии внутренних органов.
4. Синдром ломкой хромосомы (синдром Fra, Мартина-Белла) (q99.2)
Больных мальчиков в 2-3 раза больше, чем девочек, Мальчики болеют тяжелее. Популяционная частота 1:2 000 - 1:5 000 всех живорожденных, что сопоставимо с синдромом Дауна.
По своей природе заболевание относится к группе моногенных наследственных болезней. Но в результате генной мутации (экспансия нестабильных тринуклеотидных повторов (CGG) в 5-нетранслируемой области FMR-I гена (fragile mental retardation) значительно удлинняется соответствующий сегмент Х-хромосомы (q27.3), в результате нарушается ее структура - участок внешне напоминает »спутник» длинного плеча.
Ранее считалось, что заболевание наследуется по Х-сцепленному рецессивному типу. Женщин-носительниц в популяции 1:500 и больше. Впервые описано в 1943 г. как синдром умственной отсталости.
Минимальные диагностические признаки:
Умеренная или глубокая умственная отсталость,
Большие оттопыренные ушные раковины, выступающий лоб, массивный подбородок,
Макроорхизм,
Характерен интеллектуальный дефект:
С сфере общения - отсутствие контакта с окружающими, сверстниками;
Синдром двигательной расторможенности;
Характерны навязчивые движения (скрип зубами, раскачивание и др.);
Речь - стереотипные штампы, аграмматизм, эхолалия, неологизмы;
Агрессивность в поведении (желание делать все назло, ударить, укусить и т.п.);
Отсутствие желания понравиться, добиться похвалы, одобрения;
Аутизм, психологическая изоляция;
Отсутствие критики в поведении.
Встречаются ожирение, гинекомастия, гипоспадия, мягкая растяжимая кожа, слабость связочного аппарата коленных и голеностопных суставов, пролапс митрального клапана.
МОНОГЕННЫЕ БОЛЕЗНИ (мутации)
Моногенные синдромы и болезни (МБ) или генные (так их называют за рубежом) заболевания подчиняются менделевскому наследованию, в их основе лежат единичные генные или точковые мутации. МБ составляют значительную долю наследственной патологии и насчитывают сегодня более 4500 заболеваний. По данным литературы, в разных странах они выявляются у 30-65 детей в расчете на 1000 новорожденных, что составляет 3,0-6,5%, а в структуре общей смертности детей до 5 лет на их долю приходится 10-14%.
Многие МБ, несмотря на достаточно высокий уровень медико-биологических знаний, представляют значительные трудности в своевременной диагностике и эффективном лечении и часто приводят к значительному нарушению качества жизни больных, инвалидизации и раннему летальному исходу.
Заболевания многочислены и отличаются выраженным клиническим полиморфизмом. В основу классификации моногенных болезней положено несколько принципов:
1. По ведущей системной патологии - по органному и системному типу.
2. По этиологии. В этом случае выделяют 2 класса заболеваний:
Болезни с установленным первичным молекулярным (биохимическим) дефектом. Число таких болезней продолжает неуклонно расти: если по каталогу Маккьюсика в 1990 г. этот класс составил около 100 болезней, то на конец 1993 г. их было уже 328, а к 2001 г. насчитывалось около 500 (10-11% всех МБ);
Болезни с неустановленным первичным молекулярным (биохимическим) дефектом. На эти заболевания приходится около 90% всех МБ.
Первичный молекулярный дефект подразумевает определение дефектного гена и установления вида его конкретных изменений, появление и передача которых по наследству определяет развитие болезни, т.е. речь идет об изменениях в генетическом локусе.
Первичный биохимический дефект подразумевает уровень простой биохимической реакции (функции), в осуществлении которой участвует белковый продукт нормального гена и которая первично нарушается из-за соответствующнго дефекта в структуре белка.
3. По типу наследования патологического признака (см. в разделе «Методы мед. генетики»):
Аутосомно-доминантные (Д),
Аутосомно-рецессивные (Р),
Сцепленные с половой хромосомой (Х-сц) доминантные и рецессивные
Митохондриальные.
4. По преимущественному поражению того или иного вида обмена. Благодаря этому принципу многие МБ называются наследственными болезнями обмена веществ (НБО). Среди них выделено более 700 форм, в том числе 200 с установленным биохимическим дефектом. Среди НБО выделяют:
4.1болезни аминокислотного обмена (ФКУ, тирозиноз, алкаптонурия, лейциноз и др.);
4.2болезни углеводного обмена (галактоземия, гликогенозы, мукополисахаридозы);
4.3болезни липидного обмена (эссенциальные семейные липидозы, ганглиозидозы, сфинголипидозы, цереброзидозы, лейкодистрофии, гиперлипидемии и др.);
4.4болезни биосинтеза кортикостероидов (адрено-генитальный синдром, гипоальдостеронизм и др.);
4.5 болезни пуринового и пиримидинового обмена (оротовая ацидурия, подагра и др.);
4.6 болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглера-Найяра, порфирии и др.);
4.7 болезни эритрона (анемия Фанкони, гемолитические анемии, дефицит глюкозо-6-фосфатдегидрогеназы и др.);
4.8 болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный пери-одический паралич и др.);
4.9 болезни транспорта систем почек (болезнь де Тони-Дебре-Фанкони, витамин D-резистентный рахит, тубулопатии и др.);
4.10 болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз и др.).
Кроме того, в рамках НБО отдельно выделяют болезни накопления (тезаурисмозы). Эта группа заболеваний, вызываемых недостатком лизосомальных ферментов и проявляющихся прогрессирующим отложением веществ определенного типа (обычно предшественники реакций) в клетках различных тканей - гликогенозы, цереброзидозы и др. (описаны ниже).
Болезни аминокислотного обмена
Характерным почти для всех заболеваний этой группы является тип наследования - аутосомно-рецессивный, т.е. 25% потомства двух клинически здоровых носителей (гетерозигот) оказывается пораженным.
Общий биохимический признак - ацидоз тканей и аминоацидурия (согласно этому симптому называется вся группа заболеваний).
Неспецифические клинические признаки: рвота, обезвоживание организма (интоксикационный синдром), неврологические нарушения - летаргическое состояние или возбуждение, судорожный синдром.
С возрастом появляется задержка психомоторного развития, регрессия приобретенных ранее навыков, умственная отсталость (вплоть до идиотии) и задержка физического развития.
Фенилкетонурия (ФКУ) (E70.1)
(12q24.1 РАН, PKU1 или - в случае дефицита дигидроптеридинредуктазы - 4q15.1) Частота 1:10 000. Р.
Ребенок рождается здоровым. Фенотипические признаки - светлые волосы, светлая кожа, голубые глаза. Клинические симптомы ФКУ (умственная отсталость, судорожный синдром, гиперкинезы, походка, поза «портного», склонность к дерматитам) проявляются через 3-6 месяцев после рождения.
Основной биохимический маркер ФКУ - увеличение плазменной концентрации фенилаланина (гиперфенилаланинемия) - определяется через 3-4 дня после начала кормления.
Биохимические диагностические критерии:
Проба Феллинга (скрининг-тест): моча зеленого цвета;
Индикаторные бумажные тесты с применением биофана Р;
Тест Гатри;
Иммунно-ферментный метод на аппарате «Флюроскоп».
Уровень фенилаланина в плазме выше 200 мг/л;
Нормальный уровень в плазме тирозина;
Повышенный уровень в моче метаболитов фенилаланина (фенилпировиноградная и гидроксифенилуксусная кислоты), мышиный запах мочи;
Снижение толерантности к полученному внутрь фенилаланину;
Нормальная толерантность кофактора тетрагидробиоптерина.
Вовремя начатое лечение (диетотерапия) обеспечивает хороший клинический эффект, нормальную продолжительность жизни.
Лейциноз (болезнь кленового сиропа) (E71.0)
(19q13.1, 1p31.2 - тип 2, 6p22.2 - тип 3) Частота 1:90 000 - 120 000. Р.
Клинически заболевание проявляется на первой неделе жизни рвотой, пронзительным криком и появлением характерного запаха мочи, напоминающего запах кленового сиропа или отвара овощей. При этом появляется неврологическая симптоматика: отсутствие сухожильных рефлексов, мышечная гипотония, генерализованные и очаговые судороги, нарушение ритма дыхания. Отмечается замедленное психо-моторное развитие, в дальнейшем - умственная отсталость. Возможно развитие коматозного состояния, ранний летальный исход.
В основе заболевания - ферментный блок декарбоксилирования аминокислот с разветвленной цепью - лейцина, изолейцина, валина. Уровень этих АМК в крови и моче повышен. Повышена экскреция с мочой кетокислот.
Альбинизм.
Распространенный наследственный дефект пигментации, часто обусловленный дефектом синтеза фермента тирозиназы, необходимой для нормального меланинобразования. В сочетании с поражением различных органов и систем формирует несколько наследственных синдромов (Ципрковского-Марголиса, Ваарденбурга-Клейна, Чедиака-Хигаси).
Альбинизм тотальный (глазо-кожный) (E70.3) Частота 1:39 000. Р.
Тотальная депигментация кожи, волос, глаз, причем окраска одинакова для всех расовых групп и не меняется с возрастом. Кожа не загорает, совершенно отсутствуют невусы, какие-либо пигментные пятна. Сильно выражены нистагм, светобоязнь, красный зрачковый рефлекс. Острота зрения значительно снижена и с возрастом не улучшается. Снижена резистентность к инфекциям. Возможны эпилепсия, бесплодие. Предрасположенность к раку кожи.
Традиционные методы лечение альбинизма неэффективны. Рекомендуется использовать различные средства защиты от ультрафиолетовых лучей.
Наследственные болезни, связанные с нарушением липидного обмена
1. Болезни накопления липидов (тезаурисмозы) - внутриклеточные липидозы, при которых наблюдается преимущественное поражение клеток мозга и вторично в процесс вовлекаются проводящие пути.
Амавротические идиотии (E75.4) - группа липидозов, характеризующихся наличием в разных возрастных периодах следующих общих признаков: прогрессирующее снижение зрения; развитие деменции; спаcтические параличи; симптом «вишневой косточки» - вишнево-красное пятно на сетчатке глаза.
1. Врожденная форма Нормана-Вуда;
2. Раннедетская (инфантильная) форма Тея-Сакса (15q23-24 - HEXA).
Частота - 1:360 000 для большинства популяций и 1:3600 для евреев-ашкенази;
3. Позднедетская форма Бильмовского-Янского;
4. Юношеская форма Баттена-Шпильмейера Фогта-Шегрена;
5. Поздняя форма Куфса.
Группы риска по развитию гиперлипопротеидемии:
Семьи, где:
1) Родственники с молодых лет страдали сердечно-сосудистыми заболеваниями.
2) Встречается вегето-сосудистая дистония.
3) Встречается кожный ксантоматоз (бугорки желто-охряного цвета, плоские, от булавочной головки до 1,5 - 2 см в диаметре, сливаются).
Диагностика гиперлипопротеидемии:
1. Определение липидов (метод хроматографии).
2. Определение холестерина, триглицеридов, липопротеидов.
3. Метод ультрацентрифугирования.
4. Метод иммунохимический.
Болезни, связанные с нарушением обмена углеводов
Гетерогенная группа наследственных болезней, обусловленных различными видами нарушения обмена углеводов. Выделяют несколько групп:
Нарушения обмена моно- и дисахаридов;
Болезни накопления - гликогенозы;
Патология соединительной ткани - мукополисахаридозы;
a. Галактоземия (E74.2)
(9р13.1) Частота 1:35 000 - 50 000. Р.
Нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, нервной системы и другие органы.
Клиника:
Желудочно-кишечные расстройства - понос и рвота с первых дней жизни ребенка. После чайно-водной паузы состояние улучшается, но введение молока обуславливает рецидив нарушений со стороны ЖКТ.
Проявления недостаточности печени: стойкая желтуха с преобладанием в крови прямого билирубина, увеличение печени, цирроз печени.
Помутнение хрусталика - катаракта (появляется позднее).
Задержка психофизического развития.
Поражение почек - протеинурия, гипераминоацидурия.
b. Гликогенозы (E74.0)
Группа наследственных болезней обмена полисахаридов, развивающихся в результате нарушения синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях. Известно 11 типов гликогенозов, в основном наследуемых по аутосомно-рецессивному типу, кроме IX (Х-сцепленный рецессивный).
Клиническая картина гликогенозов характеризуется гипогликемией (рвота, судороги, потеря сознания, кома). Течение болезни зависит от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму, мышечный синдром (включая сердечную форму). Преобладание у новорожденного ребенка симптомов гипогликемии может привести к синдрому внезапной смерти. Прогноз зависит от типа болезни.
c. Мукополисахаридозы (E76.3)
Мукополисахаридозы(МПС) - гетерогенная группа заболеваний, отнесенных к наследственным болезням обмена сложных сахаров. МПС сопровождаются избыточным накоплением в тканях и повышенной экскреции гликозаминогликанов (ГАГ) - кислых мукополисахаридов, соединенных с белком и состоящих из уроновых кислот, аминосазароз и нейтральных сахаров. Указанные комплексы существуют в форме протеогликанов, являющихся важнейшими компонентами основного структурного белка волос (0-кератин) и структурного белка соединительной ткани (коллаген).
Для большинства МПС характерен аутосомно-рецессивный тип наследования, кроме синдрома Хантера (Х-сцепленный рецессивный).
Клиническая картина: манифестация болезни, как правило, в возрасте до 7 лет, задержка роста (до карликовости), контрактуры суставов, кифоз/кифосколиоз/сколиоз, массивный череп с глубоким и удлинненым турецким седлом, короткая шея, деформация грудной клетки, веслообразные ребра, укорочение трубчатых костей, грубые черты лица, помутнение роговицы, гепатоспленомегалия, задержка психического развития, судороги, глухота, грыжи (пупочная, паховая, паховомошоночная), врожденные пороки сердца, гликозамино-гликанурия (100-200 мг в сутки). Характерные для МПС признаки дисморфизма получили название Гаргоилический фенотип (Гаргоилизм - синоним МПС).
Диагностика МПС основывается на совокупности данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5-10 раз). Точная идентификация типов МПС возможна только с помощью определения активности лизосомных гидролаз в лимфоцитах и лейкоцитах крови, культуре фибробластов кожи, биоптатов печени, а также в моче.
Лечение больных МПС в основном симптоматическое, заключается в назначении терапии, способствующей нормализации или стабилизации патологического процесса в опорно-двигательном аппарате, сердечно-сосудистой и центральной нервной системе, паренхиматозных органах, органах зрения и слуха. Перспективным считается плазмаферез.
СИНДРОМ МАЛЬДИГЕСТИИ И МАЛЬАБСОРБЦИИ.
Нутритивный статус и связанные с ним темпы физического и психо-моторного развития ребенка особенно важны в период новорожденности и раннего возраста. Различные морфо-функциональные нарушения желудочно-кишечного тракта служат причиной развития у ребенка так называемого синдрома мальдигестии и мальабсорбции.
Мальдигестия - это недостаточность пищеварительной функции ЖКТ - полостного пищеварения (синдром мальдигестии, интестинальные энзимопатии).
Мальабсорбция - это недостаточность всасывательной функции кишечника - пристеночного пищеварения (синдром мальабсорбции).
Традиционно оба синдрома объединяются понятием «синдром мальабсорбции», так как в обоих случаях на определенной стадии развивается общее нарушение пищеварительной и всасывательной функции различных отделов ЖКТ. Причины развития синдрома разнообразны: кишечные инфекции, отравления, алиментарные нарушения, аллергические заболевания и т.д. В этиологической структуре важное место занимают наследственные нарушения морфо-функционального состояния ЖКТ. Раннее выявление у ребенка наследственных заболеваний с синдромом мальабсорбции и своевременное назначение адекватной диеты и лечения - это возможность нормального физического и психо-моторного развития, профилактика инвалидизации.
Классификация наследственно обусловленных и врожденных нарушений кишечного всасывания:
1. Первичные нарушения всасывания моносахаридов:
2. Первичные нарушения всасывания аминокислот:
3. Первичные нарушения всасывания жиров:
4. Первичные нарушения всасывания витаминов:
5. Первичные нарушения всасывания минеральных веществ:
6. Первичные нарушения всасывания электролитов:
Наиболее распространенными наследственными заболеваниями, протекающими с развитием синдрома мальабсорбции, являются целиакия, дисахаридазная недоста-точность, экссудативная энтеропатия, муковисцидоз (кишечная и смешанная формы).
Целиакия (болезнь Ги-Гертера-Гейбнера, кишечный инфантилизм,
глютеновая энтеропатия, нетропическая спру) (K90.4)
Возникновение целиакии обусловлено непереносимостью компонентов белка злаковых - проламина и глютенина (общее название - глютен). В различных злаках проламины имеют различное название: в пшенице - глиадин, во ржи - секалинин, в ячмене - гордеин, в овсе - авенин и др. Наиболее высокая концентрация проламинов определяется в пшенице, ячмене и ржи.
Исследователями приводятся различные данные о частоте встречаемости целиакии: в России - 1:5 000 - 1:10 000 детей, в странах Европы 1:300 и даже 1:100 обследованных. По данным некоторых авторов девочки болеют чаще.
Целиакия наследуется по аутосомно-доминантному типу, в то же время является и полигенным заболеванием. Выявлена основная ассоциация целиакии с локусами гена главного комплекса гистосовместимости, расположенного на хромосомах 14 и 6. Проводится дальнейшая детализация молекулярного дефекта.
Патогенез заболевания до конца не выяснен. Основная схема - аутоиммунное воспаление слизистой двенадцатиперстной кишки, вызванное повреждающим действием глютена на энтероциты с дальнейшим подавлением экзокринной функции отделов ЖКТ.
Клиническая симптоматика заболевания отличается большим полиморфизмом и зависит от возраста пациента. «Классичесакая» форма целиакии проявляется клинически через 4-8 недель после введения в питание глютенсодержащих прикормов или докорма (манная каша, геркулесовые отвары). Провоцирующими факторами могут служить кишечные инфекции, ОРВИ, стрессовые ситуации. Основными симптомами являются:
Обильный, зловонный, светлый или «разноцветный», плохо отмывающийся стул 2 и более раз в сутки (80%), причем подобный стул может отмечаться периодически в течение жизни;
Увеличение размера живота (77%), определяемое по индексу Андронеску (% отношение окружности живота к росту). Нормальные значения составляют 50-52% у детей в возрасте до 1,5 лет и 41-42% - у детей старше 2 лет;
Боли в животе (77%), с локализацией в околопупочной области, чаще нарастание болевого синдрома происходит через 3-5 часов после приема пищи, характер болей - от тупых разлитых до острых. Боли проходят самостоятельно или после дефекации;
Рвота с различной кратностью - от редкой до ежедневной (47%);
Различные варианты дисфагии (89%);
Отставание массы тела и роста от основных показателей (60%);
Проявления сопутствующей пищевой аллергии - атопический дерматит (60%),
респираторный аллергоз (33%;
Проявления фосфорно-кальциевой недостаточности - боли в костях, ночные и при физической нагрузке, переломы костей при неадекватной травме, поражение зубной эмали, кариес (50%);
Раздражительность, агрессивное поведение, неспокойный сон (63%);
Кроме основных проявлений, отмечаются общие симптомы витаминно-минеральной и белковой недостаточности: частые ОРВИ, мышечная слабость, парестезии, судорожный синдром (вплоть до эпилепсии), обмороки, дистрофические изменения волос и ногтей, фолликулярный гиперкератоз, хейлиты, стоматиты, повышенная кровоточивость, гипопротеинемические отеки.
Течение целиакии характеризуется чередованием периодов обострения и ремиссии. Обострения могут протекать с прогрессирующим вовлечением в патологический процесс новых органов и систем.
Основные этапы диагностики:
1) клинический - сочетание 3 основных симптомов и 2 и более дополнительных - подозрение на целиакию.
2) лабораторный - повышение уровня антиглиадиновых антител (АГА) классов А и G - целиакия возможна с большой степенью вероятности.
3) инструментальный - выявление атрофии слизистой оболочки 12-перстной кишки, визуально и характерные морфологические признаки - диагноз целиакии подтвержден;
4) при невозможности проведения второго и третьего этапов - пробная безглютеновая диета не менее 3 месяцев, а при снижении массо-ростовых показателей - не менее года.
Основным методом лечения целиакии является, бесспорно, пожизненная диета с полным исключением всех продуктов, содержащих глютен ячменя, овса, пшеницы, ржи: хлеб белый и черный, макаронные и мучные изделия, мороженое, некоторые йогурты, импортные сыры, колбасные изделия (из-за возможности добавления муки), консервы, соусы, кетчуп, уксус (кроме яблочного), майонез, кондитерские изделия, растворимый кофе, красители, консерванты. Взрослым больным запрещается прием алкоголя и курение (из-за опасности развития опухолевых заболеваний). Недопустимыми для больных целиакией являются продукты с содержанием глютена более 1 мг на 100 г продукта (50 г пшеничного хлеба соодержит 2-3 г глютена). Разрешаются изделия из рисовой, кукурузной муки, картофельного крахмала, а также рисовая, кукурузная, пшенная, гречневая каши.
Прогноз заболевания благоприятный при соблюдении диеты.
Муковисцидоз (кистофиброз) (E84.9)
Наследственное заболевание, обусловленное системной дисфункцией экзокринных желез. Одно из тяжелейших заболеваний детского возраста.
Ген локализован в сегменте 7q32 и кодирует белок - регулятор трансмембранной проводимости (CFTR). Частота для стран Европы и Северной Америки 1:2 000; в азиатских странах встречается редко. Тип наследования - аутосомно-рецессивный.
В настоящее время известно около 900 мутаций, из которых в Европе и России наиболее часто встречается мутации F508 (потеря аминокислоты фенилаланина в позиции 508).
Подобное изменение структуры ДНК приводит к нарушению функции белка, обеспечивающего транспорт иона хлора через апикальную часть мембраны эпителиальной клетки. Вследствие этого дефекта анионы хлора задерживаются в клетке, усиливают абсорбцию катионов натрия и воды, «высушивая слизь», продуцируемую экзокринными железами. Увеличение вязкости секрета приводит к закупориванию протоков экзокринных желез, накоплению его и образованию кист - развивается картина системной дисфункции экзокринных желез.
Поражаются те органы, в эпителиальных клетках которых нарушена функция хлоридных каналов. Это верхние и нижние дыхательные пути, потовыводящие протоки, выводные протоки слюнных желез, поджелудочной железы, желчевыводящие пути, кишечник, семявыносящие протоки у мальчиков.
Выделяют следующие клинические формы:
Смешанная (70% случаев);
Преимущественно легочная (11%). При этом поражение органов пищеварения
минимально или отсутствует;
Абортивная или стертая (11%);
Преимущественно кишечная (5%);
Мекониальная непроходимость (3%).
Это деление условно, так как одна форма практически всегда переходит в другую.
Поражения поджелудочной железы, вызванное закупориванием её протоков густым, вязким секретом, приводит к образованию кист и в дальнейшем к кистозно-фиброзному перерождению паренхимы поджелудочной железы. В более старшем возрасте из-за фиброзного перерождения стромы поджелудочной железы и при поражении островков Лангерганса развивается эндокринная недостаточность поджелудочной железы, приводящая к формированию сахарного диабета. Поражение желчевыводящих протоков приводит к развитию билиарного цирроза с портальной гипертензией (выражающейся в варикозном расширении вен пищевода), асцитом, спленомегалией и гиперспленизмом. У всех больных МВ при УЗИ можно выявить картину холестаза, на этом фоне у 15% больных развивается желче-каменная болезнь.
Из-за нарушенного транспорта ионов натрия и хлора содержание электролитов и жидкости в просвете кишечника резко снижено, что может привести к мекониальной кишечной непроходимости в младенчестве, выпадению прямой кишки в детстве, развитию кишечной непроходимости на уровне илеоцекального угла в более старшем возрасте.
Двусторонняя атрезия семявыносящих протоков приводит к аноспермии и мужскому бесплодию.
Больные МВ страдают синуситами, у 30% больных выявляется полипоз носа.
Патологический процесс в легких начинается после рождения ребенка, когда в просвете бронхов формируется густой и вязкий секрет, приводящий к нарушению мукоцилиарного клиренса. Возникающий мукостаз является благоприятной основой для развития инфекционно-воспалительных процессов. Хронический гнойный бронхит, частые бронхопневмонии приводят к формированию ателектазов, бронхиоло-бронхоэктазов, в более позднем возрасте возникают осложнения в виде пневмоторакса, легочного кровотечения. По мере прогрессирования патологических изменений в бронхолегочной системе нарастает вентиляционно-перфузионный дисбаланс, возникает гипоксия, легочная гипертензия и формируется хроническое легочное сердце.
Классическая диагностическая триада при МВ:
1. Положительный потовый тест (хлориды пота); после стимуляции потоотделения пилокарпином с помощью ионофореза пот собирается на фильтровальную бумагу (не менее 100 мл) и проводится его химический анализ с определением концентрации натрия и хлора. Положительным тестом считается превышение концентрации в 60 ммоль/л у детей и 70 ммоль/л у взрослых.
2. Рецидивирующая легочная патология инфекционно-воспалительного характера.
3. Кишечный синдром.
Диагноз подтверждается:
1. Отягощенным семейным анамнезом;
2. Положительным результатом генетического анализа;
3. Повышенным содержанием иммунореактивного трипсина в крови;
4. При определении сниженной активности ферментов поджелудочной железы в кале (протеолитическая активность кала) - копрологический анализ;
4. Азооспермией, вызванной обструкцией семявыносящих протоков.
Помимо перечисленных диагностических тестов, разработаны программы массового скрининга новорожденных, включающие два этапа:
первый этап - определение иммунореактивного трипсина в пятнах крови и альбумина в меконии;
второй этап - определение электролитов в поте.
Лечение муковисцидоза в основном симптоматическое, целью проводимой терапии являются:
1. Уменьшение бронхиальной обструкции. Проводится при помощи:
Муколитиков - препаратов, уменьшающих вязкость мокроты (N-ацетилцистеин, лазолван и др.).
Бронходилататоров - - агонистов (сальбутамол, сальметерол), М-холинолитиков (ипратропиума-бромид), теофиллинов.
Используется ДНКаза - препарат, расщепляющий ДНК разрушенных нейтрофилов в просвете бронхов.
Кинезитерапия - это вид физиотерапии, направленный на эвакуацию мокроты из бронхиального дерева. Формы кинезитерапии: цикл активного дыхания, аутогенный дренаж, постуральный дренаж в сочетании с перкуссионным массажем.
2. Борьба с инфекцией. Антибактериальная терапия при МВ опирается на определение чувствительности к антибиотикам выделенного возбудителя.
3. Улучшение нутритивного статуса больного:
Постоянная заместительная терапия ферментными препаратами нового поколения (микросферические ферменты с рН-чувствительной оболочкой (креон, панцитрат).
В диете не должно быть ограничений. Калораж питания должен составлять 120-150% от необходимого, 35% из них за счет жиров.
Дополнимтельный прием витаминов А, Д, Е, К.
4. Оперативное лечение - трансплантация - производится пересадка обоих легких, возможна также пересадка комплекса « легкие-сердце».
5. Генная терапия является новым этапом в лечении МВ. Синтезирован ген белка МВТР, проводятся активные попытки ввести этот ген в эпителиальные клетки бронхов. Проведены клинические испытания с использованием вектора аденовируса и липосомами.
Врожденные пороки развития.
Врожденный порок развития - возникшее внутриутробно стойкое морфологическое изменение органа, системы органов, части тела или всего организма, выходящее за пределы вариаций строения и нарушающее его (её) функцию. Пороки развития, не сопровождающиеся функциональными нарушениями чаще называют врожденными аномалиями (например, деформации ушных раковин - не обезображивающие лица больного и существенно не отражающиеся на восприятии звуков).
К врожденным порокам относятся следующие нарушения развития:
АГЕНЕЗИЯ - полное врожденное отсуствие органа.
АПЛАЗИЯ - врожденное отсутствие органа с наличием его сосудистой ножки.
ВРОЖДЕННАЯ ГИПОПЛАЗИЯ - недоразвитие органа, проявляющееся дефицитом относительной массы или размеров органа, превышающих отклонение в две сигмы от средних показателей для этого возраста.
ВРОЖДЕННАЯ ГИПОТРОФИЯ - уменьшение массы тела плода или новорожденного. У детей старшего возраста применяют термин «нанизм» (карликовость, микросомия, наносомия).
ВРОЖДЕННАЯ ГИПЕРТРОФИЯ - увеличенная относительная масса (или размеры) органа за счет увеличения количества (гиперплазия) или объема (гипертрофия) клеток.
МАКРОСОМИЯ (гигантизм) - увеличенная длина тела.
ГЕТЕРОТОПИЯ - наличие клеток, тканей или целых участков органа в другом органе или в тех же зонах того же органа, где их быть не должно.
ГЕТЕРОПЛАЗИЯ - нарушение дифференцировки отдельных типов ткани.
ЭКТОПИЯ - смещение органа, т.е. расположение его в необычном месте.
УДВОЕНИЕ - увеличение в числе того или другого органа или его части. Часто используется частица «поли-» (полидактилия).
АТРЕЗИЯ - полное отсутствие канала или естественного отверстия.
СТЕНОЗ - сужение канала или отверстия.
НЕРАЗДЕЛЕНИЕ (слияние) органов или двух симметричных или асимметрично развитых однояйцевых близнецов. Используется частица «син-» (синдактилия).
ПЕРСИСТИРОВАНИЕ - сохранение эмбриональных структур, в норме исчезающих к определенному периоду развития (открытое овальное окно или артериальный проток у ребенка старше трех месяцев). Одна из форм персистирования - дизрафия - незаращение эмбриональной щели (расщелины губы, неба, позвоночника, уретры).
ДИСХРОНИЯ - нарушение темпов (ускорение или замедление) развития.
По этиологическому признаку целесообразно различать три основные группы пороков:
В зависимости от последовательности возникновения различают:
первичные - непосредственно обусловленные воздействием тератогенного фактора (генетического или экзогенного).
вторичные - являются осложнением первичных и всегда патогенетически с ними связаны, например: атрезия водопровода мозга (первичный порок), приведшая к развитию гидроцефалии (вторичный); или spina bifida (первичный), сопровождающаяся косолапостью (вторичный). Названные гидроцефалия и косолапость могут быть и первичными пороками, возникновение их при этом будет непосредственно связано с воздействием повреждающих факторов или с генными мутациями.
По распространенности в организме первичные ВПР целесообразно подразделять на:
изолированные (одиночные, локальные) - локализованные в одном органе (например, стеноз привратника или персистирование артериального протока);
системные - пороки в пределах одной системы (например, хондродисплазия, артрогриппоз);
множественные - пороки, локализованные в органах двух и более систем.
Наиболее распространенной классификацией изолированных и системных ВПР является классификация, в основу которой положен не этиологический, а анатомо-физиологический принцип деления тела человека на системы органов. Именно по этому принципу построена классификация ВОЗ, рекомендованная для учета болезней и причин смерти, принятая XXIX Всемирной ассамблеей здравоохранения в 1975 г. Множественные ВПР целесообразно подразделять по этиологическому принципу. Таким образом, предлагается следующая
классификация ВПР:
А. Врожденные пороки развития органов и систем:
1. Пороки ЦНС и органов чувств
2. Пороки лица и шеи
3. Пороки сердечнососудистой системы
4. Пороки дыхательной системы
5. Пороки органов пищеварения
6. Пороки костно-мышечной системы
7. Пороки мочевой системы
8. Пороки половых органов
9. Пороки эндокринных желез
10. Пороки кожи и ее придатков
11. Пороки последа
12. Прочие пороки
Б. Множественные врожденные пороки:
1. Хромосомные синдромы
2. Генные синдромы
3. Синдромы, обусловленные экзогенными факторами (многофакторные)
4. Синдромы неустановленной этиологии
5. Множественные пороки неуточненные
Основные принципы взаимосвязи между воздействием на плод каких-либо факторов и формированием порока:
Специфичность тератогена. Тератогенный фактор (ТФ) вызывает возникновение специфических ВПР или пороков определенного типа.
Время воздействия ТФ. Существуют терминационные периоды для различных органов и систем, и только воздействие в этот критический период приведет к формированию ВПР соответствующего органа, системы или ряда систем, если терминационные периоды совпадают.
Доза тератогена. Для многих ТФ существует порог концентрации, ниже которого статистическая вероятность тератогенного эффекта ничтожно мала.
Генетическая конституция матери и плода во многом определяет устойчивость к воздействию ТФ (например, только у 11% матерей, принимавших во время беременности дифенилгидантоин, развился гидантоиновый синдром плода).
Выделяют тератогенные факторы биологической (инфекционной), физической и химической природы.
Среди биологических факторов значительная роль принадлежит инфекционным агентам (особенно ТОRCH -инфекциям):
Токсоплазмоз - нарушение роста плода и развития мозга;
Сифилис - нарушение роста плода, развития мозга и скелета;
Вирус краснухи - вызывает катаракту, глухоту, задержку умственного развития, ВПС;
Цитомегаловирус - нарушение роста плода, аномалии со стороны ЦНС, иногда только
потеря слуха;
Вирус герпеса - обычно не вызывает пороков, но при пренатальном заражении может
привести к развитию энцефалита новорожденных.
Химические вещества и медицинские препараты:
Алкоголь - нарушает рост плода, приводит к развитию аномалий головного мозга, лицевого дисморфизма, ВПС (у 30-40% детей от матерей, часто употребляющих алкоголь во время беременности, развивается алкогольный синдром плода. Частота в популяции 1-2:1000 новорожденных).
Гидантоин - нарушение роста плода, развитие аномалий скелета и ЦНС
(гидантоиновый синдром).
Талидомид - пороки развития конечностей и расщелины неба (талидомидный синдром).
Ретиноевая кислота - ВПР головного мозга, уха и сердца.
Тетрациклин - образование темных пигментных пятен на поверхности зубов.
Варфарин - кровотечения, атрофии зрительной системы (варфариновый синдром).
Другие препараты - антиконвульсанты, антикоагулянты, антитиреоидные препараты,
химиопрепараты, йодсодержащие вещества, свинец, литий, ртуть, противозачаточные
препараты.
Радиационные воздействия - ТФ, который может вызывать ВПС, нарушая деление клеток и органогенез. Преимущественно поражается нервная система и череп (микро- и гидроцефалия), глаза (катаракта, колобома).
Метаболические нарушения у матери:
При сахарном диабете - 10-15% риск возникновения у детей ВПР сердца, скелета,ЦНС.
Основной ТФ - гипергликемия.
При фенилкетонурии - почти всегда формируются ВПС и дефекты ЦНС.
Основной ТФ - избыточная концентрация метаболитов фенилаланина.
Механические воздействия на плод:
Внутриматочные (неправильное анатомическое строение матки, внутриматочные опухоли или фибромы) - ограничивают движения и рост плода, что может привести к развитию ягодичного предлежания, деформаций лица, вывиха бедра, косолапости. При маловодии может возникнуть гипоплазия легких, деформации лица и др. аномалии (синдром Поттер).
Внешние - способствуют развитию нарушений кровоснабжения плода, образованию складок амниотического мешка (амниотические сращения - тяжи Симонара), результатом чего может стать гипоплазия конечностей или поперечные ампутации (амниотические перетяжки).
Лекция №9.
Мультифакториальные заболевания
Мультифакториальные заболевания (наследственно предрасположенные, много-факторные, «Сomplex genetic disorders») - это большая и нозологически разнообразная группа болезней, развитие которых определяется взаимодействием определенных наследственных факторов (мутаций или сочетаний аллелей) и факторов среды. Этиология и патогенез данных болезней сложны, многоступенчаты и во многом еще неясны и, естественно, разные для каждой болезни.
Болезни с наследственной предрасположенностью возникают у лиц с соответствующим генотипом (сочетание «предрасполагающих» аллелей) при провоцирующем действии факторов среды,
С определенной долей условности мультифакториальные болезни можно разделить на:
1) врожденные пороки развития,
2) распространенные психические и нервные болезни,
3) распространенные болезни «среднего» возраста.
ВПР мультифакториальной природы - расщелина губы и неба, спинно-мозговая грыжа, стеноз привратника, анэнцефалия и черепно-мозговая грыжа, вывих бедра, гидроцефалия, гипоспадия, косолапость.
Бронхиальная астма
Распространенность - от 4 до 8% среди всего населения, в детской популяции - до 10%.
Бронхиальная астма - заболевание, в основе которого лежит хроническое аллергическое воспаление бронхов, сопровождающееся их гиперреактивностью и периодически возникающими приступами затрудненного дыхания или удущья в результате распространенной бронхиальной обструкции, обусловленной бронхоконстрикцией, гиперсекрецией слизи, отеком стенки бронхов.
Основные предрасполагающие факторы - атопия и гиперреактивность бронхов - генетически обусловлены. Последние данные свидетельствуют о том, что три группы признаков (уровень специфического IgE, уровень общего IgE и наличие бронхиальной гиперреактивности) наследуются независимо друг от друга. Гены, предопределяющие продукцию специфических IgE, локализованы на коротком плече 11 хромосомы (11q13), связаны с аллелями II класса HLA. Контроль базального уровня общего IgE осуществляется кластером генов длинного плеча 5 хромосомы (5q31.1). Бронхиальная гиперреактивность связана с генетическими маркерами того же сегмента (5q31.1- q33). На этом же участке расположены гены интерлейкинов (IL-4,IL- 9 и др.), активирующих тучные клетки, ген, кодирующий 2-адренорецептор.
Каждый из генетических факторов предрасположенности повышает вероятность заболевания астмой, а их комбинация приводит к высокому риску реализации болезни при минимальном участии факторов внешней среды. Наиболее значимые из них - патологическое течение внутриутробного периода, недоношенность, нерациональное питание, поллютанты и табачный дым, ОРВИ.
Часто БА сочетается с атопическим дерматитом, основным предрасполагающим фактором которого также является атопия. Риск развития атопического заболевания у детей (независимо от формы) составляет 60-80%, если оба родителя больны и/или имеют отягощенную наследственность; до 50% и выше - по линии матери; 25-30% - по линии отца.
Язвенная болезнь
Язвенная болезнь - хроническое рецидивирующее заболевание, характеризующееся образованием язвы желудка или двенадцатиперстной кишки вследствии нарушения общих и местных механизмов нервной и гуморальной регуляции основных функций гастродуоденальной системы и трофики, а также развития протеолиза слизистой оболочки.
С генетических позиций язвенную болезнь можно разделить на четыре основные группы:
1. Язвенная болезнь в целом как болезнь с наследственным предрасполо-жением,характерным для мультифакториального наследования.
2. Язвенная болезнь, укладывающаяся в моногенный (чаще аутосомно-доминантный) тип наследования.
3. Язвенная болезнь как одно из клинических проявлений нескольких наследственных синдромов.
4. Язвенное поражение гастродуоденальной системы при некоторых соматических заболеваниях.
Сахарный диабет
Сахарный диабет - гетерогенное по своей природе заболевание, в этиологии и патогенезе которого участвуют как внутренние (генетические, иммунные), так и внешние (вирусные инфекции, интоксикации), факторы, взаимодействие которых приводит к нарушению углеводного обмена.
Роль генетических факторов в развитии сахарного диабета:
1. Сахарный диабет, равно как и нарушенная толерантность к глюкозе, является постоянным компонентом примерно 45 наследственных синдромов.
2. Различные клинические проявления и распространенность сахарного диабета в этнических группах не всегда объясняются только различиями в условиях внешней среды.
3. Среди больных сахарным диабетом есть группы людей с разной зависимостью от инсулина.
4. Существует сахарный диабет взрослых, который наследуется моногенно по аутосомно-доминантному типу.
5. Различные варианты сахарного диабета можно моделировать на экспериментальных животных.
На развитие сахарного диабета влияет мутация одного или нескольких генов. Формирование патологического фенотипа, т.е. развитие клинических проявлений сахарного диабета при наличии наследственной предрасположенности происходит при обязательном участии факторов внешней среды. В этиологии сахарного диабета большое значение имеют различные стрессовые факторы, инфекции, травмы, операции. Для инсулинзависимого сахарного диабета факторами риска являются некоторые вирусные инфекции (краснуха, ветряная оспа, эпид.паротит, вирус коксаки, эпид.гепатит), токсические вещества. Для инсулиннезависимого сахарного диабета факторами риска являются избыточная масса тела, отягощенная по сахарному диабету наследственность, атеросклероз, артериальная гипертензия, дислипопротеинемия, снижение физической активности, несбалансированное питание.
Группы высокого риска по сахарному диабету:
1. Монозиготный близнец больного сахарным диабетом;
2. Лицо, у которых один или оба родителя больны или болели сахарным диабетом;
3. Женщина, родившая ребенка с массой тела более 4,5 кг., а также мертвого ребенка с гиперплазией островкового аппарата поджелудочной железы.
Нерациональная лекарственная терапия - один из важнейших факторов риска развития сахарного диабета.
Препараты, действующие на углеводный обмен: адреналин, аминазин, кофеин, сальбутамол, суросемид, кортикостероиды, тироксин, СТГ, АКТГ, допегит, клофелин, трентал, ПАСК, салицилаты, бутадион, сульфаниламиды.
Наследственные синдромы, сопровождающиеся нарушением толерантности к глюкозе или резистентностью к инсулину:
Генные: Синдром Луи-Бар, муковисцидоз, анемия Фанкони, дефицит глюкозо-6-фосфат дегидрогеназы, гликогеноз I типа, подагра, гемохроматоз, хорея Гентингтона, синдром Лоуренса-Муна-Барде-Бидля, синдром Прадера-Вилли.
Хромосомные: синдром Дауна, синдром Клайнфельтера, синдром Шерешевского-Тернера.
Ишемическая болезнь сердца (ИБС)
ИБС возникает вследствие уменьшения или прекращения снабжения миокарда кровью в связи с патологическим процессом в коронарных сосудах. Основная часть ИБС представляет собой мультифакториальную патологию, характеризующуюся формированием заболевания в процессе взаимодействия генетических и средовых факторов, которые приводят к непосредственным причинам ИБС: I) спазм коронарных артерий; 2) атеросклероз коронарных сосудов. Основной патофизиологический механизм ИБС - несоответствие между потребностью миокарда в кислороде и возможностями коронарного кровотока их удовлетворить.
К генетически детерминированным факторам риска по ИБС относятся:
Пол пробанда: у женщин клинические проявления возникают на 10-15 лет позже, это связано с гормональными различиями и морфологическими особенностями строения коллатеральных сосудов коронарных артерий;
Тип телосложения: чаще сердечно-сосудистые заболевания, связанные с атеро-склерозом, встречаются у лиц с гиперстеническим типом телосложения;
Личностные особенности: описан тип личности «А» (энергичность, ускоренный темп работы, стремление к достижению поставленных целей, люди эмоциональны, подвержены стрессовым факторам), при котором частота ИБС наблюдается в 2 раза чаще, чем при типе «В».
Определенное строение коронарных сосудов;
Повышенный уровень общего холестерина в крови; высокий уровень в крови липопротеидов низкой и очень низкой плотности (ЛПНП и ЛПОНП); низкая концентрация липопротеидов высокой плотности (ЛПВП);
Небольшая активность рецепторов ЛПНП;
Нарушения в свертывающей системе крови (увеличение фибриногена в сыворотке крови, наследственная недостаточность фибринолитической активности);
Артериальная гипертензия;
Сахарный диабет.
Фенокопии гиперлипидемий, связанные с нарушением липидного обмена, могут быть обусловлены действием таких средовых факторов, как:
Курение (смертность от ИБС среди курящих выше в 2-5 раз, чем среди некурящих);
Гиподинамия (риск смерти от ИБС у физически малоактивных людей в 3 раза выше);
Несбалансированное питание; изменение минерального состава воды - длительный прием мягкой воды, бедной минеральными солями (Ca, Мg, литий, цинк);
Воздействие отрицательных психосоциальных факторов;
Прием контрацептивных стероидов
СЕМИОТИКА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Семиотика
Семиотика - учение о знаках. Семиотика наследственных болезней - учение о симптомах болезней, правильном обозначении их круга, морфологических и функциональных изменениях органов и частей тела, динамике клинических проявлений, т.е. это необходимое условие для успешной диагностики заболевания.
Огромное разнообразие наследственных заболеваний, синдромов, пороков развития характеризуется различными сочетаниями отдельных признаков (симптомов), общее число которых, по некоторым оценкам, превышает три тысячи. По четкости регистрации они подразделяются на три группы:
1. альтернативные: либо есть, либо нет (примеры - преаурикулярные папилломы, шейные фистулы, четырехпальцевая складка ладони и т.д.);
2. измерительные: признаки, определяемые абсолютным или относительным количественным значением (удлиннение, укорочение, увеличение, уменьшение и др., примеры - арахнодактилия, брахидактилия, макро- и микроцефалия и т. д.);
3. описательные: признаки, характеризующиеся изменениями кожи, волос, мягких тканей и др., к которым трудноприменимы количественные оценки. В отличие от признаков первой группы они требуют в своем обозначении сравнительных характеристик (примеры - пятна на коже цвета «кофе с молоком», паклеобразные волосы, клювовидный нос, воронкообразная грудная клетка и т. д.).
Патологический фенотип определенного наследственного синдрома складывается из более или менее устойчивого сочетания отдельных симптомов (минимальные диагностические признаки), создающих в совокупности специфическое «фенотипическое ядро» заболевания, являющееся основой для установления диагноза.
Синдром - совокупность внешних и внутренних, морфологических и функциональных аномалий и врожденных пороков, вызванных единым морфологическим фактором.
Обычно тот или иной синдром имеет от 1-2 до 5 (редко более) соответствующих признаков. Задача врача состоит в том, что бы увидеть данные аномалии и правильно их интерпретировать. Сложность заключается в том, что нередко отсутствует параллелизм между значимостью (в смысле тяжести) симптома для пациента и его диагностической ценности (информативности) - в смысле возможности установления диагноза. Так, например, в случае синдрома Аарскога (лице-пальце-генитальный синдром) основным поводом для обращения является задержка роста, нередко сочетающаяся с крипторхизмом, - достаточно широко распространенные состояния, а диагностически значимым (высокоинформативным) симптомом данного синдрома является необычная форма мошонки, окружающая в виде валика основание полового члена ребенка (шалевидная мошонка) - вполне безобидный признак. При синдроме Ваарденбурга основной жалобой является снижение слуха (вариант врожденной нейро-сенсорной тугоухости за счет гипоплазии Кортиева органа), а основой установления диагноза являются обнаруживаемые на волосистой части головы и лице малые аномалии: седая прядь волос, аномально короткие глазные щели за счет латерального смещения внутренних углов глаз (телекант), медиально расширяющиеся брови с тенденцией к сращению на переносьи (синофриз), гетерохромия радужных оболочек, широкий корень носа. И подобных примеров можно привести множество.
Наряду с высокоинформативными симптомами в структуре наследственных синдромов обычно присутствуют и фоновые признаки: симптомы, часто встречающиеся при многих наследственных синдромах (а также и в общей популяции), создающие в своей совокупности фон диспластичного развития ребенка (стигмы дизэмбриогенеза - это небольшие отклонения, которые не сказываются существенно на функции органа и не уродуют внешность больного): эпикант, деформация ушных раковин, высокое небо, измененная дерматоглифика, клинодактилия, различные варианты синдактилий и т.д. Диагностическая значимость отдельно взятого признака этой группы относительно невелика, однако недооценивать их также не следует, особенно, когда к ребенку есть более серьезный повод для «претензий» в виде задержки физического, интелектуального и полового развития и т. д. Обнаружение двух и более (в отечественной педиатрии - 7-10) малых аномалий (стигм) у больного служит показанием для проведения тщательного клинического обследования.
Клинико-морфологическое обследование (паспортная диагностика) пациента предполагает определенную последовательность, примерная схема которго представлена ниже (т.н. карта фенотипа).
Генетические заболевания не всегда удается диагностировать на ранних этапах. Некоторые патологии начинают проявляться только после рождения малыша. Так, синдром кошачьего крика можно определить по характерным изменениям .
Синдром кошачьего крика – что это такое?
Синдром Лежена, синдром кошачьего крика – генетическое заболевание, которое связано с нарушением структуры хромосомы. Чаще изменениям подвергается пятая хромосома. Развитие синдрома напрямую связано с изменением структуры ее короткого плеча. При патологии наблюдается частичная потеря данного участка (1/2 или 1/3 длины) или полное отсутствие.
Впервые патология была описана в 1963 г. Изучением проблемы занимался французский педиатр, генетик Ж. Лежен. Впоследствии заболевание начали именовать его фамилией – синдром Лежена. В медицинской литературе чаще встречается название синдром кошачьего крика – согласно основному, специфическому признаку патологии.
Синдром кошачьего крика у детей
Данное заболевание относят к редким генетическим патологиям. Синдром Лежена (фото новорожденных приведено ниже) легко определить по характерным изменениям внешности малыша, поэтому диагностировать патологию врачам удается сразу после появления малыша на свет. Однако для постановки диагноза проводят полное обследование.
Как утверждают специалисты, появление характерного плача, напоминающего мяуканье кошки, связано с особенностями строения гортани. Она имеет явное недоразвитие, узкая, а хрящи, ее образующие, тонкие. В 30% случаев дети с синдромом Лежена теряют эту особенность развития к трем годам жизни, однако у некоторых она остается до смерти.
Синдром кошачьего крика – симптомы
Понять, что такое синдром кошачьего крика, можно, просто взглянув на малыша с данной патологией. В большинстве случаев такие дети имеют малую массу тела, даже при рождении в срок. Большинство малышей с данной патологией рождаются раньше положенного времени. Средняя масса тела таких новорожденных не превышает 2,5 кг. При этом беременность может протекать без осложнений – многие будущие мамы узнают о генетическом заболевании ребенка только после родов.
Главным характерным признаком заболевания является плач, напоминающий кошачье мяуканье. Он имеет высокий диапазон, пронзительный. Связано это с узким просветом гортани, малым размером надгортанника. При осмотре горла обнаруживается необычная складчатость слизистой оболочки, мягкая консистенция хрящей. Определить заболевание можно и по характерным изменениям внешности ребенка:
- лицевая часть черепа преобладает над мозговой;
- лицо в форме Луны;
- антимонголоидный разрез глаз;
- деформация ушных раковин;
- плоская спинка носа;
- короткая шея.
При детальном обследовании ребенка наблюдаются следующие признаки синдрома кошачьего крика:
- снижение тонуса мышц;
- низкая рефлекторная деятельность (нарушение глотания и сосания).

Синдром Лежена – причины
Наследственное заболевание «синдром кошачьего крика» является следствием мутации 5-й хромосомы. Причины изменения генного аппарата настолько многочисленны и разнообразны, что зачастую не удается установить конкретный фактор, приведший к патологии. Как показали исследования, негативное влияние на оплодотворенную яйцеклетку происходит на этапе деления и образования зиготы. В отдельных случаях генетический материал родителей изначально является поврежденным, что и вызывает патологию у ребенка. Среди факторов, провоцирующих нарушение:
- наследственность;
- никотин;
- алкоголь;
- наркотики;
- длительное применение некоторых лекарственных препаратов (особенно в первом триместре беременности);
- ионизирующее излучение.
Синдром кошачьего крика – диагностика и лечение
Определить заболевание можно по характерным проявлениям патологии. Однако необходимо учитывать, что синдром кошачьего крика – это генетическая патология, поэтому для постановки диагноза необходимо исследование генома пациента. Первые признаки патологии могут быть обнаружены уже при УЗИ в ходе пренатального скрининга. В таком случае для подтверждения предположений используют дополнительные методы инвазивной диагностики:
- (исследование околоплодных вод);
- биопсию ворсин хориона.
При диагностировании патологии после родов диагноз ставится на основании характерного плача, изменений внешности малыша. Однако предположения врачей всегда должны подтверждаться результатом цитогенетического анализа. Данное исследование помогает установить характерные изменения генного материала и исключить другие хромосомные патологии.
Синдром кошачьего крика – кариограмма
Болезнь синдром кошачьего крика можно установить с помощью исследования кариотипа. В ходе анализа врачи внимательно изучают образец генетического материала. О наличии патологии можно говорить, когда наблюдается хромосомная аберрация в форме утраты 1/3 части хромосомы. Данным изменениям подвергается пятая хромосома.
При этом важен не размер утраченной части хромосомы, а само отсутствие данного участка. Потеря малого участка в области 5p15.2 приводит к появлению основных клинических признаков, кроме кошачьего крика. Критическим для возникновения характерного крика является выпадение участка в области 5p15.3.

Синдром кошачьего крика – частота встречаемости
Синдром кошачьего крика у человека встречается крайне редко. Как показывает статистика, данное генетическое заболевание регистрируется у 1 из 45–50 000 новорожденных. При этом прослеживается разница частоты развития патологии у мальчиков и девочек. Согласно наблюдениям, соотношение количества заболевших девочек к мальчикам с таким синдромом составляет 4:3.
Синдром кошачьего крика – продолжительность жизни
Когда у ребенка диагностируют синдром кошачьего крика, сколько живут такие малыши и каковы прогнозы – родители интересуются часто. Каждый случай индивидуален. На продолжительность и качество жизни детей с патологией влияют сопутствующие пороки и патологии внутренних органов. Зачастую прогноз неблагоприятный. Уровень психомоторного развития детей не превышает дошкольный. Около 90 % всех малышей с патологией не доживают до 10 лет, хотя известны случаи, когда пациенты с таким синдромом доживали до 50 лет.
Генетические заболевания все чаще дают про себя знать современному миру. Такая ситуация обусловлена плохой экологией, частыми стрессами беременной женщины, нездоровым образом жизни родителей или наследственностью. «Поломки» в генах приводят к различным болезням, которые зачастую проявляются сразу после рождения или еще с момента зачатия. Одним из таких заболеваний считается синдром кошачьего крика.
Что такое синдром кошачьего крика?
Синдром кошачьего крика - это генетическая болезнь, которая проявляет себя из-за нарушения структуры плеча пятой хромосомы. Данную генетическую патологию также называют синдромом Лежена. Вместо привычного плача ребенок издает тонкие высокие звуки, напоминающие кошачий крик или мяуканье. Такой звук обусловлен патологией гортани и мягкостью хрящей в этой зоне.
Это заболевание представляет собой измененную структуру пятой хромосомы. Если сказать точнее, то зачастую данная хромосома просто теряет треть или половину своей длины. Синдром кошачьего крика также может развиться, когда пятая хромосома полностью теряет свое короткое плечо, но это происходит гораздо реже.
Информация Синдром кошачьего крика часто называют редким генетическим заболеванием. На 45-50 тысяч новорожденных детей приобрести эту болезнь может только один ребенок. Как показывает статистика, чаще болезнью Лежена болеют девочки.
Специфический звук во время плача – это только одно из проявлений данного синдрома. Точно диагностировать синдром кошачьего крика можно только после цитогенетического обследования. Помочь миру бороться с данной генетической патологией помог французский педиатр Ж. Лежен. Он первый, кто в 1963 году полностью описал картину болезни.
Причины синдрома Лежена
Проявление синдрома кошачьего крика обусловлено потерей генетической информации, которая хранится на участке пятой хромосомы. В зависимости от утерянного участка отличается и симптоматика, но причины появления этой патологии зачастую одинаковые:
- случайная мутация (85-90% случаев);
- наследственность (10-15% случаев);
- возраст матери (старше 40 лет);
- употребление , наркотиков;
- проживание в загрязненной местности;
- влияние ионизирующей радиации,
- прием запрещенных химических веществ или медикаментозных препаратов.
Чем старше возраст женщины, тем выше вероятность повреждения яйцеклетки, но чаще возрастает риски у тех, чей возраст превысил 40-45 лет.
Когда женщина во время беременности употребляет алкоголь или курит сигареты, то, прежде всего, вредные вещества воздействуют на половые клетки и здоровье малыша в утробе. Наркотические вещества молниеносно разрушают генетическую структуру клеток плода.
Дополнительно Прием тяжелых химических веществ или прием сильнодействующих препаратов во время первого триместра также может послужить причиной появления синдрома Лежена. Генетические изменения могут начать формироваться еще в период оплодотворения яйцеклетки, поэтому воздействие радиации является одной из основных причин негативного воздействия на половые клетки. Подвергаются мутациям клетки и матери, и отца.
Генетическая предрасположенность к синдрому кошачьего крика занимает первенство в этом списке. Если у родителей один ребенок уже болеет этим синдромом, то второй ребенок с вероятностью в 50% также может заболеть.
Признаки и симптомы кошачьего крика у детей
Самым ярким симптомом, который может говорить о наличии синдрома кошачьего крика, является тонкий пронзающий плач малыша. Он напоминает тонкое мяуканье кошки или ее крик. Ученые выяснили, что такой звук появляется из-за дефектов анатомического строения гортани. Она отличается узким просветом. Слизистая оболочка гортани имеет нехарактерную складчатость, а хрящи долгое время имеют мягкую структуру.
Установить точный диагноз врачам помогает не только характерный плач малыша, ведь этот симптом может вообще пропасть у трети детей до двух лет.
У некоторых детей кошачий крик может стать хроническим симптомом, поэтому медики обращают внимание сразу на целый спектр изменений развития ребенка, а именно:
- сильное слюноотделение,
- малый вес при рождении (до 2,5 кг),
- пороки сердца,
- нарушения рефлексов глотания и сосания,
- запоры,
- агрессивность поведения,
- однообразие в движениях,
- плохое физическое развитие,
- низкое качество речи,
- гиперактивность.
Кроме вышеперечисленных симптомов, врачи обращают внимание и на внешние изменения ребенка, которые с каждым годом все ярче себя проявляют. Чаще всего видоизменяется форма лица и глаз, поэтому также нужно обращать внимание на следующие признаки:
- лунообразная форма лица;
- широко посаженные глаза;
- плоская переносица;
- широкий нос;
- косоглазие;
- небольшой размер черепа (микроцефалия);
- выпячивание лобных бугров;
- низко посаженные уши;
- аномалии прикуса;
- короткая шея.
У больного с синдромом Лежена могут наблюдаться сопутствующие патологии, но они не всегда входят в перечень обязательной симптоматики:
- чрезмерная гибкость суставов;
- болезни сердца и сосудов;
- проблемы с опорно-двигательным аппаратом и ЖКТ;
- различные болезни глаз.
Подростки с синдромом кошачьего крика зачастую не имеют проблем в репродуктивной сфере. Половое созревание проходит вовремя. У девочек период первых менструаций не отличается от здоровых детей, но иногда может встречаться двуроговость матки. У мальчиков редко отмечают маленький размер яичек, но качество спермы нарушается мало.
Родителям необходимо терпимо переносить все эмоциональные срывы таких детей, ведь их поведение часто отличается истерией и агрессией к окружающему миру. Психотерапевты часто диагностируют глубокую отсталость в умственном развитии и идиотию.
Кариотип синдрома кошачьего крика
Проводить врачи намерены проводить только в тех случаях, если есть явные факторы риска. Кариотипирование – это анализ крови, который позволяет точно изучить ядро клеток обоих родителей. После взятия крови из нее выделяют отдельные клетки, которые в дальнейшем будут проходить специальное окрашивание. За счет этой методики врачам проще определить хромосомы.

Кариотип у матери должен быть – 46, ХХ, а у отца - 46,ХY. После определения кариотипа проще определить наличие или отсутствие отклонений.
Если есть малейшие отклонения от нормы, то вероятность появления у ребенка синдрома кошачьего крика существенно возрастает. При этом ученые определили, что явных изменений у родителей может и не наблюдаться. С точностью определить предрасположенность ребенка к какому-то генетическому нарушению нельзя.
Диагностика
Проводить диагностику синдрома кошачьего крика и в принципе генетических мутаций принято в два этапа. Первый этап заключается в общем обследовании женщин. Цель обследования - выявить повышенный риск вынашивания плода с патологиями хромосом. Второй этап диагностики отвечает за окончательное подтверждение диагноза.
Все исследования должны проводиться только в специализированных медицинских центрах перенатальной диагностики. Зачастую все исследования осуществляются еще на этапе беременности.
Узнать о наличии или отсутствии синдрома кошачьего крика можно еще в период первого триместра беременности.
Чтобы с высокой точностью установить этот диагноз Лежена, врачам нужно провести несколько исследований:
- сбор анамнеза,
- проверку кариотипа родителей,
- инвазивные исследования,
- исследования крови по определенным показателям,
- послеродовая диагностика.
Анамнез представляет собой простую беседу родителей с генетиком или педиатром. Ответственные родители проходят этот этап еще в период . Чтобы максимально обезопасить своего будущего малыша от различных генетических болезней, родителям желательно проходить все необходимые исследования.
Дополнительно Если у родителей есть высокая предрасположенность к синдрому кошачьего крика, то стоит задуматься о зачатии ребенка. Такое заключение должно поступить только от лечащего врача, но родители всегда оставляют право выбора за собой.
Кариотипирование родителей – это простой тест крови на наличие любых генетических отклонений, которые могут повлиять на развитие плода.
Уже на период беременности посмотреть на развитие плода, но даже на этом этапе врач не может точно диагностировать синдром Лежена. Если во время диагностики была замечена угроза хромосомной аномалии, то возможно повторное УЗИ.
Анализ крови на плазменные маркеры – это достаточно эффективный метод диагностики генетического заболевания у плода. Его проводят в виде анализа крови беременной женщины. Для этого собранный материал подвергают воздействию плазменных маркеров хромосомных болезней.
Определить точное наличие синдрома Лежена методом плазменных маркеров не получится, так как это анализ просто диагностирует наличие генетической патологии.
Инвазивные исследования отличаются наибольшей точностью, так как они с вероятностью в 98-99% могут определить наличие какой-либо мутации генов у плода. Проводят такую диагностику еще в первом триместре беременности. Для этого осуществляют взятие ткани у плода. После установки точного диагноза женщина вправе сама решать – сохранять или прерывать беременность.
Когда ребенок с синдромом кошачьего крика уже появился на свет, его здоровье продолжают проверять. Своевременная диагностика сопутствующих болезней на ранних сроках позволяет повысить его выживание в будущем. Для этого ребенок проходит ряд исследований:
- осмотр неонатолога и педиатра,
- рентген или УЗИ брюшной полости,
- анализ мочи и крови,
- биохимическое исследование крови по разным показателям.
После точной диагностики должно поступить лечение всех имеющихся симптомов. Жизненные функции ребенка должны поддерживать различные терапии, а иногда и хирургические операции.
Лечение синдрома кошачьего крика
Прежде всего, лечение синдрома Лежена начинается к консультации узкопрофильных врачей. Для этого нужно пройти обследование у:
- гастроэнтеролога,
- офтальмолога,
- кардиолога,
- ортопеда,
- лора,
- и т. д.
После можно приступать к лечению имеющихся патологий. Вылечить синдром кошачьего крика современная медицина еще не может, поэтому нужно поддерживать все жизненные функции детей с таким диагнозом уже с рождения. Для этого родители должны внимательно присматриваться к своему ребенку. Врачи рекомендуют быть под постоянным наблюдением у детского невролога. Этот врач может назначать соответствующие медикаментозные препараты для стимуляции психомоторики.

Если ребенок имеет , то часто врачи прибегают к хирургическому вмешательству для устранения патологий.
Дети с таким диагнозом часто отличаются повышенным тонусом мышц, поэтому им нужно посещать кабинет массажиста и ЛФК. Для улучшения речи в старшем возрасте стоит посещать логопеда.
Прогноз синдрома Лежена
Сфера лечения хромосомных патологий стала развиваться более стремительно, но синдром кошачьего крика так и остается неизлечимым. Глядя на статистику, то приблизительно 90% всех детей с этим заболеванием не доживают до 10 лет. Бывали случаи, когда больные с синдромом Лежена жили до 50-60 лет. Делать какие-то предсказания в плане продолжительности жизни таких людей нет смысла, так как каждый человек индивидуален. Немаловажную роль играет и список сопутствующих заболеваний. Если их своевременно лечили, то продолжительность жизни может увеличиться.
Чтобы максимально продлить годы жизни людей с синдромом кошачьего крика, необходимо сразу после рождения точно определить тяжесть врожденных пороков.
Добиться хорошего результата в преодолении разных патологий опорно-двигательного аппарата помогут различные массажи и лечебные физкультуры. Только регулярные посещения врачей, своевременное лечение и диагностика сопутствующих болезней может продлить жизнь человеку с этим заболеванием.
Информация Уход за детьми с болезнью Лежена требует немалых денежных средств, поэтому часто государство открывает специальные фонды для поддержки детей с генетическими отклонениями. В некоторых случаях не помогает даже дорогостоящее лечение, так как количество пороков слишком большое. Маленькому организму тяжело справляться с такой нагрузкой. Смертность детей с таким синдромом остается очень высокой.
Если у родителей получилось улучшить состояние здоровья своего ребенка, то он в будущем сможет освоить средний уровень словарного запаса. Те слова, которые он будет знать, ребенок сможет применять в быту. Если смотреть на ситуацию с другой стороны, то уровень психомоторики останется, как у ребенка дошкольного возраста.
Синдром кошачьего крика – это неизлечимое генетическое заболевание. Будущим родителям необходимо ответственно подойти к процессу проверки собственного здоровья еще в период планирования беременности. Если в семье или роду были случаи проявления этого синдрома, то посещение генетика должно быть обязательным. Проводить диагностику данного заболевания современная медицина позволяет еще в период первого триместра беременности. Поэтому будущим родителям необходимо строго отнестись к своему образу жизни еще до зачатия ребенка.
Синдром кошачьего крика
Синдром кошачьего крика (также известен под названиями: синдром делеции короткого плеча 5 хромосомы, 5р синдром или синдром Лежена) - редкое генетическое заболевание, связанное с отсутствием части 5 хромосомы. У больных детей с этим заболеванием (преимущественно, но нельзя сказать, что у всех) проявляется плач, который похож на кошачий крик, именно поэтому этот синдром получил название Cri-Du-Chat Syndrome, что происходит от французских слов (плач кошки или крик кота). Впервые болезнь описал Жером Лежен в 1963 году. Частота возникновения синдрома - 1 ребенок на 50000 родившихся, встречается во всех этнических групп и чаще ею болеют женщины, соотношение мужской и женской стати составляет 4:3.
Признаки и симптомы
Как уже было отмечено, синдром получил свое название из-за характерного плача детей (он аналогичен мявканню котенка, крика кошки), страдающих этим заболеванием. Это крик происходит из-за проблем с гортанью и нервной системой. Около 1/3 детей теряют эту особую характерную черту до 2 лет. Другими симптомами, которые указывают на заболевания синдромом кошачьего крика являются:
проблемы с питанием в связи с трудностями при глотании и сосании;
низкий вес ребенка при рождении и низкие темпы развития (в первую очередь физического);
существенная задержка развития когнитивных, речевых функций и функций движения;
проблемы с поведением, такие как: гиперактивность, агрессия, истерики и однообразные движения, постоянно повторяются;
нетипичные черты лица, которые могут со временем исчезнуть или усилиться;
чрезмерное, неконтролируемое слюноотделение;
запоры.
Дополнительными типичными признаками заболевания можно назвать: гипотонию, микроцефалию, задержку физического развития, круглое лицо с полными щеками, гипертелоризм, эпикантус, опущенные углы глазных щелей, косоглазие, плоскую спинку носа, опущенные углы рта, микрогнатию, низко расположеные уши, короткие пальцы, 4 -х пальцевую ладонную складку и порок сердца (например, дефект межжелудочковой перегородки (вентрикулосептальный дефект), дефект межпредсердной перегородки (атриосептальный дефект), открытого артериального протока, тетраду Фалло). У людей с синдромом кошачьего крика обычно нет проблем с половой системой и репродуктивностью (рождением детей).
В позднем детстве и подростковом возрасте выводы врачей включают значительные нарушения умственного развития, микроцефалии, огрубение черт лица, надбровные дуги, глубоко посаженные глаза, гипопластическая перегородку носа, тяжелые нарушения прикуса и сколиоз. Больные девушки достигают половой зрелости, развитие вторичных половых признаков, и менструация у них возникают обычно вовремя. Строение половых органов и путей у лиц женского пола, как правило, нормальное, за исключением двурогой матки, которая местами встречается у этой группы больных. У мужчин яички часто малы, но сперматогенез преимущественно незначительно нарушен.

Синдром кошачьего крика связан с частичной делецией короткого плеча 5 хромосомы, которая также называется "5p моносомия". Примерно 90% случаев этого генетического заболевания являются результатом спорадических или случайных мутаций, то есть новые случаи. Остальные 10-15% возникают из-за неравного разделение родительского генетического материала с нарушением сбалансированной транслокации, где 5р моносомия часто сопровождается соответствующей трисомией этой части генома. Эти лица могут иметь более серьезные проявления заболевания, чем те у кого проявляются изолированные случаи моносомии с 5р (вследствие делеции).
В большинстве случаев заболевание сопровождается полной потерей дистально-расположенной генетической информации, составляющей 10-20% генетического материала на коротком плече пятой хромосомы. Менее 10% случаев имеют другие редкие цитогенетические аберрации (например, интерстициальную делецию, мозаичность, кольца и новые транслокации). Делеция хромосомы 5, родительского происхождения примерно в 80% случаев происходит заново.
Потеря небольшого участка в зоне 5p15.2 (критической для этого заболевания области) коррелирует со всеми клиническими признаками синдрома за исключением кошачьего крика, который возникает при условии нарушений в области 5p15.3 (кошачьей критической области). Полученные результаты свидетельствуют, что две несмежные критические области содержат гены, вовлеченные в этиологию этого заболевания. Два гена в этих регионах, семафорин F (SEMA5A) и дельта Катенин (CTNND2), потенциально участвуют в мозговом развитии. Удаление гена транскриптазы обратной теломеразы (hTERT) локализированной в 5p15.33 может способствовать фенотипическому изменению у больных синдромом кошачьего крика.